- 2024-06-12 15:26 9694
- 产品价格:面议
- 发货地址:北京海淀 包装说明:不限
- 产品数量:9999.00 套产品规格:不限
- 信息编号:218625240公司编号:14832749
- 王经理 微信 185101038..
- 进入店铺 在线咨询 QQ咨询 在线询价
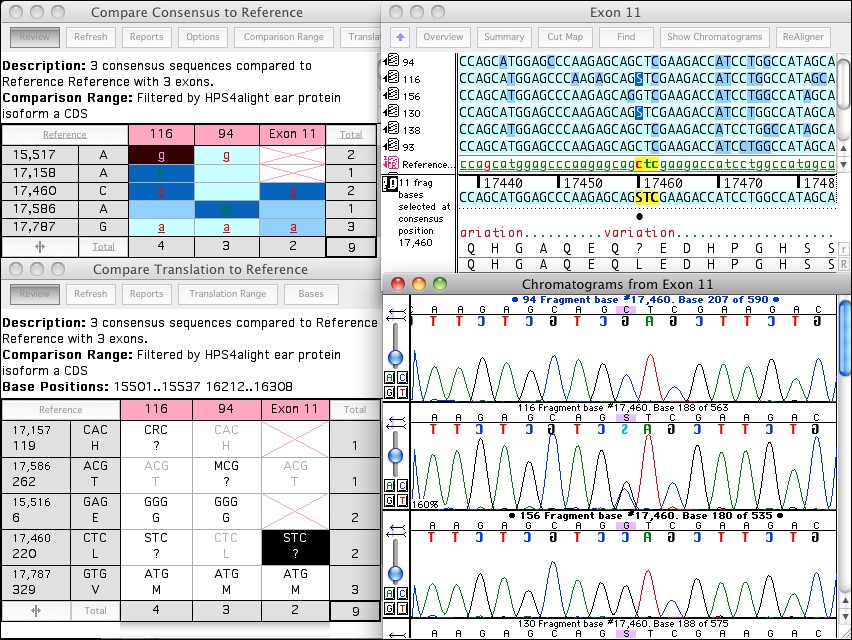
正规代理 sequencher实用教程
- 相关产品:
科学软件网提供软件和培训服务已有19年,拥有丰富的经验,提供软件产品上千款,涵盖领域包括经管,仿真,地球地理,生物化学,工程科学,排版及网络管理等。同时还有的服务,现场培训+课程,以及本地化服务。
BWA, Velvet, Maq, GSNAP and Tablet are only the start for Sequencher. If you are sequencing mixed populations, then combine the power of reference-guided-alignment with de novo assembly. Align reads to a reference and capture the unaligned reads for further rounds of reference-based alignment or de novo assembly.
For example, with the click of a button you can convert 90 files, 45 pairs of forward and reverse sequences, into 45 contigs named according to your Patient IDs. A change in your sequence assembly parameters regroups your fragments, so you can assemble the contigs according to Clone ID, Date, Primer, or any other characteristic you record in your sequence names.
MUSCLE is a command line program, which means that normally you would be using this program through a Terminal application. Sequencher gives you access to MUSCLE’s power without the problems of learning to use the UNIX command line.
Once the process is complete and MUSCLE has built the alignment, you will see the results as a contig within Sequencher. Use Sequencher’s tools to annotate your alignment or export your alignment and place it into a special phylogenetics program.
科学软件网不定期举办各类公益培训和讲座,让您有更多机会免费学习和熟悉软件。
联系电话是4008104001, 主要经营北京天演融智软件有限公司(科学软件网)主营产品PSCAD, CYME, SPSSPRO, Stata, Matlab,GAMS,Hydrus,GMS,Visual Modflow 等各学科软件,科学软件网有20多年的软件销售经验,提供专业销售和培训服务,还有更多的增值服务。目前,科学软件网提供的软件有数百种,软件涵盖的领域包括,经管,仿真,地球地理,生物化学,工程科学,排版及网络管理等各个学科。。
单位注册资金单位注册资金人民币 1000 - 5000 万元。
{kind=link}